The Atkinson Lab has a long-standing interest in the structure, function and genetics of
human and mouse complement proteins including their “abuse” by microbial pathogens. Further, the lab has made significant progress in defining how mutations in the activating proteins and their regulators drive functional consequences related to immunoinflammatory and biological debris handling human diseases.
For example, the chronic degenerative disease known as age-related macular
degeneration (AMD) afflicts more than 50 million individuals worldwide and represents the
leading cause of blindness in the elderly in developed countries. Recent studies (particularly
employing human genetic analyses as well as mouse models) defined a pathologic role in AMD for alterations in complement system genes coding for inhibitory as well as activating proteins that span the allelic spectrum from common to rare variants.
Another illness we study, atypical hemolytic uremic syndrome (aHUS), is a much rarer,
acute, and often lethal disease that most commonly occurs in early childhood. It is characterized by a triad of microangiopathic hemolytic anemia, thrombocytopenia and acute renal failure.
These two seemingly quite disparate diseases share the common theme that alterations in
complement control can lead to over exuberant, tissue damaging responses by the complement system.
See our pertinent review of this area: https://pubmed.ncbi.nlm.nih.gov/27959629/ and a 2019
paper by Java et al https://pubmed.ncbi.nlm.nih.gov/31312772/
Recent work by our lab in collaboration with that of a former fellow, Claudia Kemper
now at the National Institutes of Health, led to the discovery of an intracellular complement
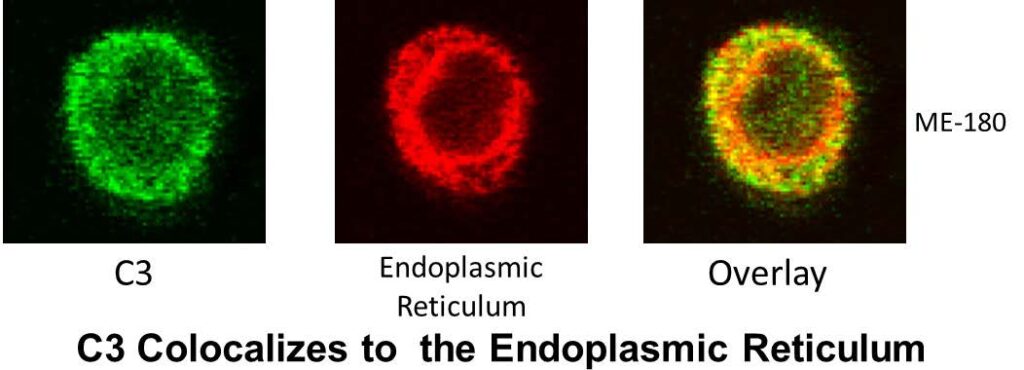
system. As demonstrated in the top figure, complement system protein C3 (and other
complement factors) is not only synthesized and secreted by the liver (traditional view) but also is stored intracellularly in the endoplasmic reticulum of most cells (new view). Prior to these studies, the complement system was considered to operate primarily in blood. However, we now appreciate that complement has an intracellular (likely the original system) arsenal of components providing both an intracellular and extracellular immune host defense, but also assisting in modulating key cellular functions (Immunity 2013; 39(6) 1143-57).
See our review: https://pubmed.ncbi.nlm.nih.gov/28196665/ and the 2019 paper by Kulkarni etal: https://pubmed.ncbi.nlm.nih.gov/30156437/
In a departure from the study of complement, we are also heavily invested in defining the
pathogenesis of and in identifying a treatment for a rare, fatal disease discovered here at
Washington University School of Medicine called RVCL-S (retinal vasculopathy with cerebral
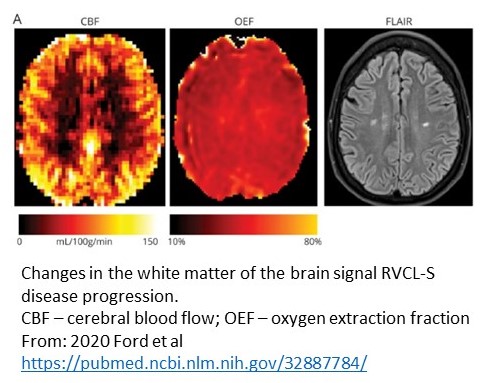
leukoencephalopathy and systemic manifestations). The autosomal dominant disease, featuring a COOH-terminal frameshift mutation in TREX1, strikes in middle-age (~35 yrs) and leads to a progressive loss of vision and brain function. We have established the first RVCL-S Center and are conducting therapeutic clinical trials for this 100% penetrant and fatal microvasculopathy.
See website: https://rvcl-research.wustl.edu/
See recent papers: Ford et al. 2020 https://pubmed.ncbi.nlm.nih.gov/32887784/ and
2021 Macaron et al https://pubmed.ncbi.nlm.nih.gov/34044261/
Atkinson Lab Research Profile